Since the 1970s, intra-aortic balloon pumps (IABPs) have been the gold standard of minimally invasive hemodynamic support, but more recently developed percutaneous ventricular assist devices (pVAD) are offering an alternative. Clinical data shows pVADs offer more support than IABPs, but their much higher price tag currently restricts their use to niche applications, usually after an IABP fails to deliver enough support.
Recent clinical data on both types of systems has not shown the sort of blockbuster efficacy data that was originally expected, instead showing IAPBs and pVADs had similar patient outcomes in most cases.
Another trend in the hemodynamic market is the use of small, lightweight extracorporeal membrane oxygenation (ECMO) systems as pVADS when high levels of support are needed in extremely sick or high-risk patients.
All of these devices reduce or eliminate the need for inotropes to pharmacologically induce the heart to beat harder. All these systems help off-load the left ventricle and perfuse organs in hemodynamically compromised patients, aiding patient survival. The main questions for hospitals looking to purchase these systems boil down to cost, ease-of-use and the level of efficacy.
Clinical Data Not as Positive as Vendors Anticipated
While IABPs have been the gold standard for hemodynamic support in low ejection fraction patients, especially those with myocardial infarction complicated by cardiogenic shock, they were first introduced prior to U.S. Food and Drug Administration (FDA) clinical trial requirements to prove efficacy. So, until now, only a few registry studies and clinical trials have shown IABPs can improve blood pressure and coronary perfusion.
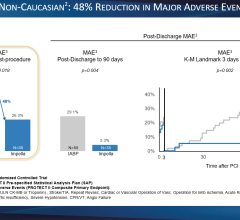
Based on this limited data, international guidelines recommend the use of IABPs for cardiogenic shock. Use of IABPs for cardiogenic shock is estimated at 25-40 percent of these cases. The IABP-SHOCK II trial, an independent investigator study supported by Maquet, was created to show benefit when using IABPs for this indication. It was expected to show IABPs reduce the 30-day mortality rate compared to standard medical therapy. But, instead of showing the superiority of IABP therapy, 30-day results from the large, randomized, multicenter trial did not meet the pre-specified 12 percent improvement in survival endpoint. Although IABPs proved to be very safe, the study provided little evidence that they are associated with hemodynamic improvement, failing to reduce rates of mortality and showing no improvement in blood pressure, no reduction in time in the intensive care unit and no improvement in organ perfusion.
“There is not a one-size-fits-all device for these patient populations, so patient selection for each type of device is very important,” said Luca Lombardi, M.D., chief medical officer, Maquet Cardiovascular. He said this was one of the problems with the SHOCK II study, because some patients received IABP therapy before and others after revascularization, and there might be more benefit to earlier use of hemodynamic support.
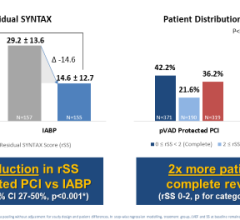
Despite the apparent advantages of pVAD therapy based on higher levels of support (2.5 to 5 lpm as opposed to about 0.5 lpm for IABPs), their much higher cost has kept usage to just a fraction of the market that IABPs dominate. Several randomized, controlled trials demonstrated a better hemodynamic profile compared to IABP, but pVAD use failed to translate into improved 30-day survival.
One highly anticipated trial was the PROTECT II trial (presented at ACC 2011), in which Abiomed hoped to show better clinical efficacy of the Impella 2.5 device over IABPs. However, after about 327 patients were enrolled, the trial was ended prematurely by the Data Safety Monitoring Board because it was clear the primary endpoint of the trial would not be met. The data showed Impella and IABPs had about the same outcomes, although some subsets showed a slight advantage for Impella. The bottom line from the trial was that efficacy was substantially similar, so there was not justification for using a much more expensive device.
Based on experience, the Impella 2.5 lpm device only delivers about 1.5 to 1.7 lpm of flow, said Alan L. Gass, M.D., medical director, heart failure, heart transplantation and mechanical circulatory support, Westchester Heart and Vascular, and associate professor of medicine, New York Medical College. For this reason, he is not sure it is worth the higher price for the Impella for only an incremental increase in the level of support. Abiomed said it has study data showing its device can achieve an average of 2.2 to 2.3 lpm.
“The cost is sky-high compared to an IABP, so I’m not sure what role it will play with only the small amount of additional support it can provide,” Gass said.
However, he said the new Impella CP device is supposed to have a higher capacity at 4 lpm, which might merit its use in specific cases where IABPs cannot deliver enough support.
Reducing STEMI Infarct Size
The FDA granted CardiacAssist Inc. an investigational device exemption (IDE) for a pivotal clinical study of the TandemHeart pVAD in December 2012. The TandemHeart to Reduce Infarct Size (TRIS) trial will evaluate the effectiveness of ventricular unloading on the reduction of infarct size for patients who have suffered a severe heart attack.
The company says its pre-clinical animal studies showed a strong correlation between high degrees of ventricular unloading and improved myocardial salvage. Patients enrolled in the TRIS trial will be randomized to receive either conventional therapy or a TandemHeart device to unload the left ventricle along with percutaneous coronary intervention (PCI). The primary efficacy endpoint of the study is improvement in myocardial salvage index (MSI). Additionally, the economic impact of TandemHeart therapy will be studied through secondary endpoints, including long-term mortality and the rates of repeat hospitalization and implantable cardioverter-defibrillator (ICD) usage.
New IABP Devices
Maquet received FDA 510(k) clearance for its new Mega 7.5 French 30 cc and 40 cc intra-aortic balloon (IAB) catheters in May 2012. The larger volume balloons displace more blood, allowing clinicians to provide patients with greater hemodynamic support, regardless of their height. The balloon uses a unique wrap and does not have a step-down between the balloon membrane and the catheter shaft, which the company says may decrease bleeding when using a sheathless approach. The new Mega IABs also provide a patented, more durable Durathane balloon membrane and a co-lumen catheter design with a large 0.027-inch inner lumen for a reliable pressure transducer signal.
Additionally, all Mega IABs come with two Statlock IAB stabilization devices, which allow the catheter to be secured to the patient’s leg without sutures.
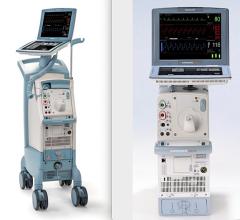
Since October 2011, Maquet has added new sizes to its higher efficacy IAB product lines to improve the delivery of hemodynamic support to the most critically ill patients. In addition to the Sensation Plus 8 French 50 cc catheter, Maquet has launched the Sensation Plus 7.5 French 40 cc IAB for patients 5 feet to 5 feet 4 inches (152 to 162 cm). In 2012, Maquet also launched Cardiosave, the company’s new fiberoptic IAB pump, which is designed with a large touch screen display and is smaller, lighter and quieter than other pumps.
New pVAD Devices
Abiomed received FDA clearance for its improved Impella CP (Cardiac Power) pVAD last fall. It is the same size as the Impella 2.5 liter-per-minute device, but delivers up to 4 liters per minute, representing about 80 percent of the blood flow in a normal heart. The Impella CP, like the Impella 2.5, is small enough for transcatheter deployment without the need for a surgical cut-down. This has been a disadvantage of the Impella 5.0 liter-per-minute device, which has a 21 French in-line pump motor, requiring a cut-down.
In November 2012, Abiomed also received FDA IDE for the use of the new Impella RP (right side percutaneous) in a pivotal clinical study in the United States. The first patient was recruited for the study in April. The Impella RP is a percutaneous heart pump that is implanted through a single access site in the patient’s leg and deployed across the right side of the heart without requiring a surgical procedure. The study, which is expected to begin in 2013, will enroll 30 patients from 10 different hospital sites and is estimated to take up to 24 months to complete. The study will enroll patients that present with signs of right-side heart failure, require hemodynamic support and are being treated in the catheterization lab or cardiac surgery suite.
The RECOVER RIGHT study will collect safety and effectiveness data on the use of the Impella RP and will be applied toward the submission of a humanitarian device exemption (HDE).
In March, Thoratec Corp. announced the first European patient to be implanted with its new HeartMate Percutaneous Heart Pump (PHP). The patient was supported for more than 60 minutes during a high-risk PCI and was hemodynamically stable during a three-vessel intervention, while the patient’s ejection fraction was less than 30 percent. Two additional patients were treated as part of this first-in-man series.
Thoratec is a leader in surgically implanted LVADs used as a bridge-to-transplant in heart failure patients, but the HeartMate PHP is a new venture into the minimally invasive cath lab space. The company purchased the technology in 2010 for $8.5 million from Swedish company Getinge AB, the parent company of IABP maker Maquet. Thoratec said the device will provide greater flow rates and a smaller size than existing devices.
The HeartMate PHP is a catheter-based heart pump designed to provide left ventricular support. Upon insertion into the femoral artery via an integrated 12 French introducer sheath, the PHP catheter is advanced into the left ventricle where the distal end of the catheter expands to 24 French, allowing for enhanced blood flow with low levels of hemolysis. Thoratec has designed the system to facilitate rapid insertion using an intuitive control console and to provide 4 to 5 lpm of mean flow.
Thoratec intends to pursue clinical investigations on the use of HeartMate PHP to support patients undergoing high-risk percutaneous interventions as well as those in cardiogenic shock, a population with consistently high mortality rates. The company is planning a continuation of the pilot study during the second quarter and anticipates initiating a CE mark study in the second half of 2013.
Another company developing a catheter pump is start-up German company Cardiobridge. The company has collected data on its first 30 patients who received the Reitan Catheter Pump (RCP). The device uses a 10 French catheter for low insertion profile, but once deployed expands out with folding propeller blades and a protective cage. Similar to an IABP, it is positioned in the decending aorta, but uses a propeller pump instead of a balloon. And unlike a balloon pump, it does not require ECG synchonization. It is designed for circulatory support for acute decompensating heart failure and high-risk PCI.
FDA Reviews IABPs and pVADs
The FDA premarket approval (PMA) and 510(k) device review process went into effect in the late 1970s and early 1980s, after IABPs were already on the market. The devices were essentially “grandfathered” as an effective medical device without the need to undergo an FDA pivotal trial to show efficacy and safety. More than 30 years later, the FDA’s Circulatory System Devices Panel decided to review IABPs. This past December, the panel voted to support the FDA’s reclassification of IABPs from the most stringent regulations as a Class III device to the less stringent Class II category. Indications that received a recommendation to be reclassified to a Class II designation include acute coronary syndrome, complications of heart failure of both ischemic and non-ischemic etiologies, and cardiac and non-cardiac surgery.
According to the FDA’s panel of physicians and representatives, this recommendation was based on 40 years of extensive clinical experience and literature, which support the hemodynamic effects and the safety and effectiveness of IABPs in the aforementioned patient populations.
Gass testified at the December FDA hearing that despite the fact that IABPs were not required to undergo a PMA study to prove safety and efficacy, they do indeed save patients’ lives and have an impeccable safety record.
“I firmly believe the IABP offers hemodynamic support,” Gass said. “In my experience, it is certainly enough to bring the patient to hemodynamic stability … The IABP is incredibly safe and systems have improved with new balloon materials and smaller sheaths. It is almost a complication-free device.”
IABP makers Maquet and Teleflex say the devices deliver about 0.5 liters per minute of augmentation flow, but Gass said his experience is that IABPs offer more support than has been tested in clinical trials. Due to the location of the balloon in the aorta, more support is delivered to vessels closest to the balloon, most importantly the kidneys. “It does a lot more than we are observing in testing,” Gass stated. He said patients in danger of their kidneys shutting down due to lack of perfusion will see immediate diuretic response. He said increased urine output is seen in patients with IABPs all the time.
“As the leaders in intra-aortic balloon therapy, we are proud that the proven nature, tremendous volume of clinical evidence and clinical utility was validated by both the FDA and members of the panel, and that IABPs have set the bar for other devices in this category,” Lombardi said. “IAB therapy has been trusted by physicians for decades, and we believe this panel vote is a testament to the importance and utility of this treatment as the standard first-line therapy for patients requiring hemodynamic support.”
The panel also recommended that IABPs retain a Class III designation for the treatment of septic shock and intraoperative pulsatile flow generation, indications for which the devices are less commonly used.
At the same meeting in December, the panel also reviewed possible reclassification of temporary ventricular support devices, which include the Impella and TandemHeart devices. The Circulatory System Devices Panel voted to retain Class III status for these devices.
In the case of the Abiomed Impella device, the panel wanted more evidence of its efficacy and required Impella to enter the FDA’s 515i program. This program reviews all previously 510(k) cleared Class III devices to determine the appropriate controls to ensure safety and effectiveness. The classification of Impella as a Class III device requires PMA controls (versus prior 510(k) Class III special controls) to ensure safety and effectiveness in the intended use.
“The data was not very impressive and the FDA panel did not feel Abiomed met any of the primary endpoints of their clinical study,” Gass said. “There is also an undercurrent thought that Abiomed only has a handful of very enthusiastic users concentrated at just a few centers.”
John Somberg, M.D., cardiologist, professor of medicine, chief, division of clinical pharmacology and director, autonomic laboratory, Rush University, Chicago, spoke at the FDA hearing and suggested Abiomed should pursue a noninferiority study relative to IABP.
“Here we have a very large study compared to what we’ve dealt with in the past, with PROTECT II looking at the IABP versus these pumps, and there really was not a significant difference. I know they were looking for superiority, but there really wasn’t,” Somberg said.
As per the 515i reclassification process and confirmation by the FDA panel, products will remain on the market while the manufacturers submit PMA applications. The panel said Abiomed’s existing data, combined with its U.S. registry, can be used as part of the data for its PMA application.
Michael R. Minogue, Abiomed president, chairman and CEO, said the PMA will strengthen the company’s existing marketing clearances and expand the indications for Impella patients.
At Abiomed’s February earnings call, Minogue said the company has had multiple conversations with the FDA. “Since the December panel meeting, we have had multiple conversations with the FDA and have recently submitted an extensive briefing document, outlining the totality of the Impella data to demonstrate safety and effectiveness …The summary includes 262 patients under FDA IDE approved protocol, 685 patients that meet the FDA clinical evidence requirements, including the Impella registry, and also references 201 publications, covering 1,877 patients.”
Comparing IABP, pVAD Devices
DAIC created comparison charts comparing the currenly available intra-aortic balloon pump (IABP) systems and percutaneous ventricular assist devices (pVAD) on the U.S. market. Visit the pVAD chart (bit.ly/ZvBAPP) or the IABP chart (bit.ly/11wYWlX).

 August 14, 2023
August 14, 2023