April 28, 2010 – About 280,000 external defibrillators used worldwide in health care facilities and public places may malfunction during attempts to rescue people in sudden cardiac arrest, the U.S. Food and Drug Administration (FDA) warned this week.
Faulty components in defibrillators manufactured by Cardiac Science Corp. of Bothell, Wash., may cause the devices to fail to properly deliver a shock. In addition to failure to deliver needed shocks, other problems with the affected models may include interruption of electrocardiography (ECG) analysis, failure to recognize electrode pads, and interference or background noise that makes the device unable to accurately analyze heart rhythm.
Recalled Models
The 14 affected models, which include automated and semi-automated devices, are:
• Powerheart models 9300A, 9300C, 9300D, 9300E, 9300P, 9390A and 9390E?
• CardioVive models 92531, 92532 and 92533?
• Nihon Kohden models 9200G and 9231 and?
• GE Responder models 2019198 and 2023440.
The FDA recommends hospitals, nursing homes and other high-risk settings obtain alternative external defibrillators and arrange for the repair or replacement of the affected defibrillators. If alternative external defibrillators are not immediately available, then FDA recommends continuing to use the affected devices if needed, because they may still deliver necessary therapy.
“The FDA is issuing this notice so that users can take the proper steps necessary to assure they have access to safe and effective defibrillators,” said Jeffrey Shuren, M.D., JD, director of the FDA’s Center for Devices and Radiological Health.
Cardiac Science recalled its Powerheart and CardioVive models, manufactured between August 2003 and August 2009, on Nov. 13, 2009. But the FDA has since learned that additional Cardiac Science models, two marketed under the Nihon Kohden name, and two marketed by GE Healthcare as GE Responder have similar problems.
Cardiac Science issued a software update for two of its Powerheart defibrillators in February 2010 and plans to issue similar software updates for other affected devices. However, FDA’s review of the updated software indicates that the software detects some, but not all, identified defects.
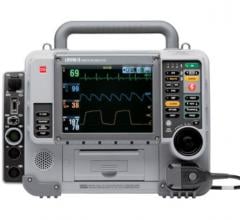
LIFEPAK 15 Recalled
A Class I recall recently announced is for Physio-Control’s Lifepak 15 Monitor/Defibrillator. These devices were manufactured between March 26 and Dec. 15, 2009 and distributed during the same time.
The reason for recall is a potential for the device to unexpectedly:
• Power off then on by itself.
• Power off then not turn on.
• Power off by itself requiring the operator to turn it back on.
• Stay powered on and not allow itself to be turned off.
Class 1 recalls are the most serious type of recall and involve situations in which there is a reasonable probability that use of these products will cause serious adverse health consequences or
For more information: www.fda.gov/Safety/MedWatch/default.htm, www.physio-control-notices.com/LP15pcba

 March 10, 2026
March 10, 2026