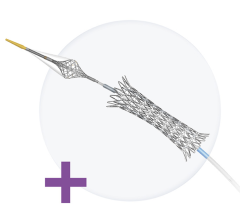
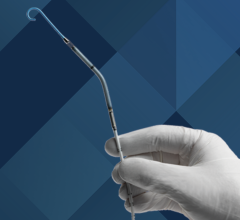
May 4, 2012 - The U.S. Food and Drug Administration (FDA) has approved the premarket approval application (PMA) for the Medinol Ltd. Presillion plus cobalt chromium (CoCr) coronary stent on a rapid exchange delivery system. This device is indicated for improving coronary luminal diameter in patients with symptomatic ischemic heart disease associated with stenotic lesions in de novo native coronary arteries (length < 30 mm) with a reference vessel diameter of 2.5 to 4 mm.
The usable length of the delivery system is 135 cm with a shaft profile of 1.9 French (64 mm) / 2.8 French (.94 mm) (proximal/distal). The catheter has a distal port (hole) approximately 25 cm from the distal tip that accesses the guidewire lumen. The guidewire lumen begins at the distal port and terminates at the distal tip. The catheter also has two markers on the proximal catheter shaft that indicate, approximately, the exit of the balloon catheter tip from the guiding catheter (brachial: 90 cm; femoral: 100 cm).
As a condition of approval, Medinol agreed to conduct two post-approval studies. It will continue follow-up of BLAST study Placebo Cohort. The prospective, observational, single-arm study will consist of continued follow up of the premarket cohort of the placebo patients from the BLAST study who will be followed annually. The individual endpoints are major adverse cardiac events (MACE), clinically driven target lesion revascularization.(TLR), target vessel failure (TVF), target lesion failure (TLF), all cause mortality, myocardial infarction (MI), composite cardiac death/Ml and stent thrombosis.
The study population will consist of the patients in the BLAST placebo cohort treated with Presillion plus CoCr coronary stent. Information on clinical outcomes will be collected annually through five years post-procedure on at least 80 percent of patients enrolled (excluding those discontinued due to death) in the BLAST clinical trial.
The company must also enroll patients in a new U.S. cohort study. It will include newly enrolled, non-randomized, multicenter, prospective, single-arm clinical study of patients treated with the Presillion plus for the treatment of de novo stenotic lesions in native coronary arteries in the U.S. population.
The primary effectiveness objective is to demonstrate that the three-year incidence of TVF (cardiac death, target vessel myocardial infarction, or clinically driven target vessel revascularization) is less than the performance goal of 33 percent derived from development of a meta-analysis of five bare metal stent trials (standard-of-care). The expected rate for TVF at three years for the Presillion plus CoCr coronary stent is 22 percent.
A secondary objective is to assess long-term safety. Secondary endpoints are all-cause mortality, cardiac death, all cause MI, target vessel MI, clinically driven TVR, acute success rates and stent thrombosis.
The study population will consist of adult patients with symptomatic ischemic heart disease due to a single de novo stenotic lesion contained within native coronary artery with reference vessel diameter between 2.5 and 4 mm and lesion length < 30 mm that is amenable to percutaneous revascularization with percutaneous coronary intervention with stent deployment.
For more information: www.accessdata.fda.gov/cdrh_docs/pdf11/p110004a.pdf
 November 24, 2025
November 24, 2025