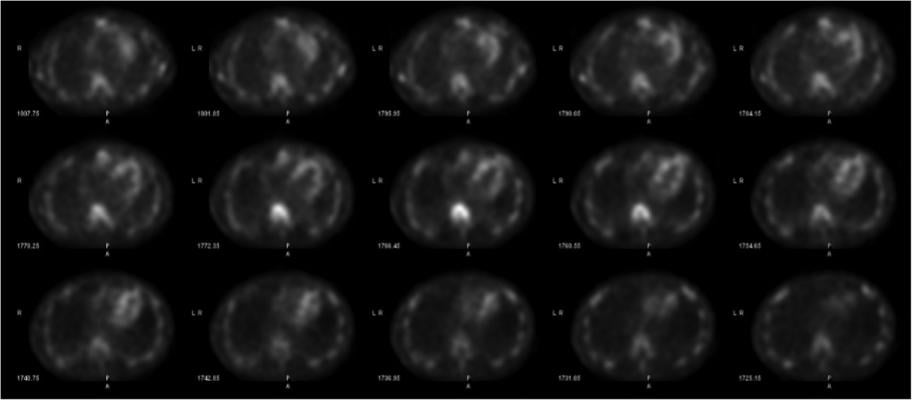
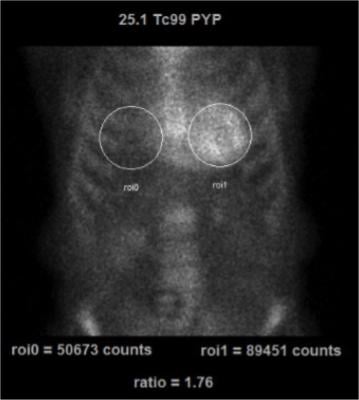
With the advent and optimization of nuclear scintigraphy protocols using bone-avid radiotracers, cardiac amyloidosis caused by transthyretin protein (ATTR) can now be diagnosed noninvasively without a costly tissue biopsy. The radiotracer 99mTc-pyrophosphate (99mTc-PYP) binds to deposited ATTR amyloid fibrils in the myocardium and can be visualized using planar and SPECT imaging. This is Figure 2, showing how SPECT imaging allows the reader to distinguish between blood pool activity (ventricular cavity, etc) and myocardial activity and identify regional myocardial differences in radiotracer uptake.
Cardiac amyloidosis is a highly morbid and underdiagnosed infiltrative cardiomyopathy that is characterized by the deposition of amyloid fibrils (misfolded protein deposits) into myocardial tissue. This results in restrictive physiology and heart failure, typically with preserved ejection fraction until late in the disease course. Cardiac amyloidosis is further characterized by the precursor proteins that ultimately develop into amyloid fibrils.
Most cases of cardiac amyloidosis are caused by amyloidogenic light chains (AL) or transthyretin protein (ATTR). AL cardiac amyloidosis is the result of misfolded monoclonal immunoglobulin light chains, which are typically found in plasma cell dyscrasias (such as multiple myeloma). ATTR amyloidosis occurs in the setting of misfolded transthyretin protein (also known as prealbumin) and has two forms: wild-type (non-mutant) or mutated transthyretin (hereditary form). Roughly 25 percent of men over the age of 80 have some degree of transthyretin myocardial deposition. However, the clinical significance of this deposition is uncertain. Heart failure symptoms typically appear once significant deposition and myocardial thickening have occurred.[1] Mutated transthyretin cardiac amyloidosis is the result of inherited mutations of the transthyretin gene, which is present in upwards of 4 percent of African-American patients.[1]
Use of MRI to Detect Cardiac Amyloidosis
ATTR cardiac amyloidosis (ATTR CA) is an increasingly-recognized, but underdiagnosed, form of heart failure. This diagnosis should be considered in patients with suggestive clinical clues such as orthostatic intolerance to typical heart failure therapies including beta-blockers and ACE-inhibitors, bilateral carpal tunnel syndrome, lumbar spinal stenosis, unexplained biceps tendon rupture, unexplained peripheral neuropathy, and low-voltage on electrocardiography. Echocardiography is typically performed and typical findings include a thickened left ventricle, diastolic dysfunction, and abnormal longitudinal strain with apical sparing. These structural findings can also be seen on cardiac magnetic resonance imaging (MRI), which provides additional tissue characterization. It shows elevated T1 signal, an expanded extra-cellular volume fraction, abnormal late gadolinium-enhancement and altered tissue kinetics with a classic failure to null the myocardium all suggest diffuse fibrosis. None of these symptom complexes or suggestive findings on echocardiography and CMR definitively diagnose cardiac amyloidosis, as they lack sufficient independent sensitivity and specificity.
Use of Biomarker Lab Testing to Confirm Amyloidosis
Confirmation of cardiac amyloidosis can be obtained invasively with a cardiac biopsy or extra-cardiac biopsy (renal, fat pad, etc.) with suggestive cardiac imaging findings or elevated cardiac biomarkers (troponin or NT-pro-BNP, in the case of AL only). Plasma cell dyscrasias associated with AL cardiac amyloidosis can be diagnosed through acquisition of serum free light chains and serum and urine immunofixation. This lab testing must be performed in any patient suspected of having cardiac amyloidosis.
Nuclear Imaging to Assess Cardiac Amyloidosis
 With the advent and optimization of nuclear scintigraphy protocols using bone-avid radiotracers, ATTR CA can now be diagnosed noninvasively without a costly tissue biopsy. This technique has moved into mainstream clinical practice. Radiotracers such as 99mTc-pyrophosphate (99mTc-PYP) in the United States and 99mTc-DPD and 99mTc-HMDP outside the U.S. bind to deposited ATTR amyloid fibrils in the myocardium and can be visualized using planar and single photon emission computed tomography (SPECT) imaging. 99mTc-PYP imaging has demonstrated high sensitivity and specificity in detecting ATTR CM in patients with biopsy-proven disease.[2] Due to its predominant uptake in ATTR CM only, likely stemming from molecular differences between the amyloid fibrils created in each of these conditions, 99mTc-PYP imaging has been shown to be effective in differentiating patients with ATTR CM from AL CM. Given the possibility of some uptake with AL CM, however, it is imperative to perform the appropriate laboratory testing to differentiate AL CA and ATTR CA prior to or after 99mTc-PYP imaging as treatments for these conditions are vastly different. In this context, 99mTc-PYP imaging plays an important noninvasive role in the evaluation of cardiac amyloidosis, which will ultimately impact prognosis, therapy, response to therapy and genetic counseling.
With the advent and optimization of nuclear scintigraphy protocols using bone-avid radiotracers, ATTR CA can now be diagnosed noninvasively without a costly tissue biopsy. This technique has moved into mainstream clinical practice. Radiotracers such as 99mTc-pyrophosphate (99mTc-PYP) in the United States and 99mTc-DPD and 99mTc-HMDP outside the U.S. bind to deposited ATTR amyloid fibrils in the myocardium and can be visualized using planar and single photon emission computed tomography (SPECT) imaging. 99mTc-PYP imaging has demonstrated high sensitivity and specificity in detecting ATTR CM in patients with biopsy-proven disease.[2] Due to its predominant uptake in ATTR CM only, likely stemming from molecular differences between the amyloid fibrils created in each of these conditions, 99mTc-PYP imaging has been shown to be effective in differentiating patients with ATTR CM from AL CM. Given the possibility of some uptake with AL CM, however, it is imperative to perform the appropriate laboratory testing to differentiate AL CA and ATTR CA prior to or after 99mTc-PYP imaging as treatments for these conditions are vastly different. In this context, 99mTc-PYP imaging plays an important noninvasive role in the evaluation of cardiac amyloidosis, which will ultimately impact prognosis, therapy, response to therapy and genetic counseling.
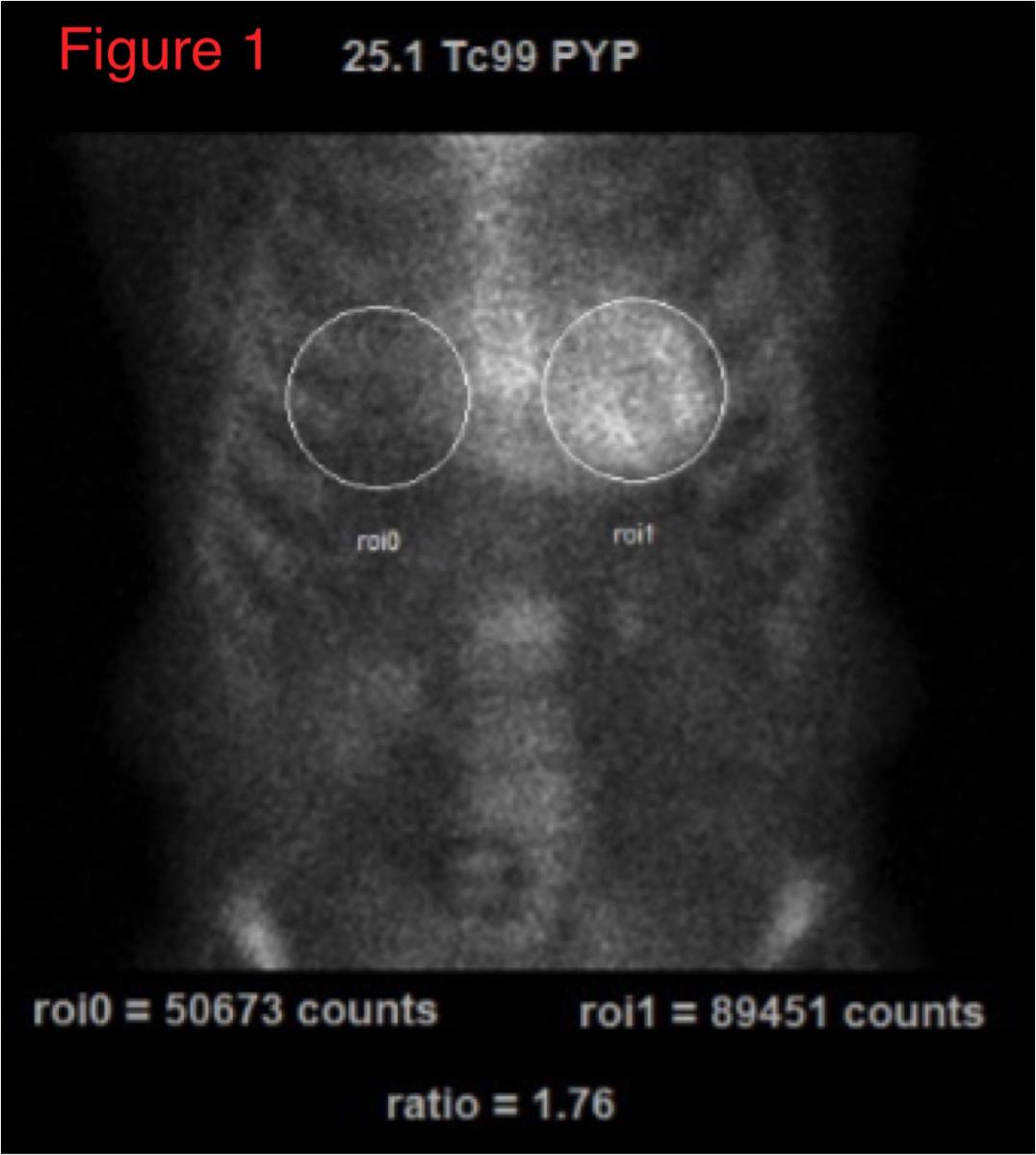
99mTc-PYP imaging can be easily performed in most clinical nuclear laboratories. The 99mTc-PYP radiotracer is readily available and prepared by commercial radiopharmacies. The imaging protocol is also relatively straightforward. Planar imaging is obtained either one or three hours post-radiotracer injection. SPECT reconstruction is necessary to confirm localization of the radiotracer in the myocardium. In some patients (renal failure, etc), significant blood pool activity can be noted and imaging is repeated at a later time point to allow adequate blood pool clearance. Planar imaging serves as a useful technique to visually interpret the degree of myocardial uptake. Quantitative analysis can be performed using planar images as regions of interest (ROI) are drawn over the heart and compared to the contralateral chest to measure the mean and relative counts (99mTc-PYP uptake).
An uptake ratio of ≥1.5 is considered diagnostic of ATTR CM (Figure 1).
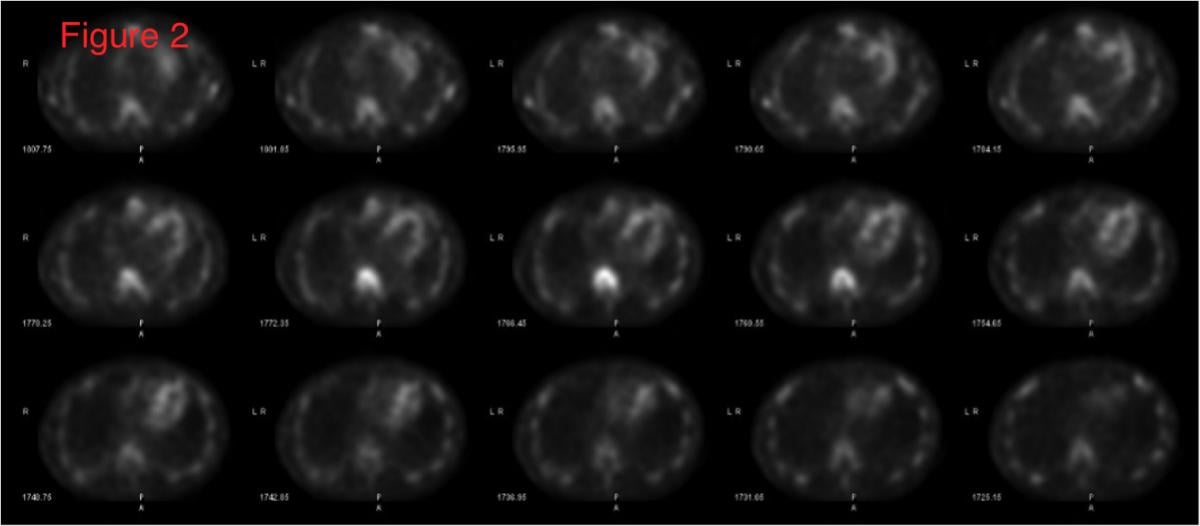 SPECT imaging allows the reader to distinguish between blood pool activity (ventricular cavity, etc) and myocardial activity and identify regional myocardial differences in uptake (Figure 2).
SPECT imaging allows the reader to distinguish between blood pool activity (ventricular cavity, etc) and myocardial activity and identify regional myocardial differences in uptake (Figure 2).
Semi-quantitative scoring is based on myocardial 99mTc-PYP uptake and is graded 0-3: 0=absent cardiac uptake, 1 equals mild uptake less than bone, 2 equals moderate uptake equal to bone, 3 equals high uptake greater than bone.[2] Based on either or both quantitative and semiquantitative analysis, the degree of uptake is evaluated as: (1) not suggestive of ATTR CA, (2) strongly suggestive of ATTR CA, or (3) equivocal for ATTR CA.
Therapy for ATTR Cardiac Amyloidosis
Noninvasive techniques to diagnose ATTR CA will only continue to become more important as treatments are now available. In May 2019, Pfizer’s Vyndamax (tafamidis) and Vyndaqel (tafamidis meglumine) became the first-ever U.S. Food and Drug Administration (FDA) approved therapies for ATTR CM. Several other investigational agents for ATTR CA are also likely to be released in the near future. These therapies will be important options for patients with few past therapeutic options. Candidacy and coverage for these therapies will likely require formal appropriate diagnosis of ATTR CM to confine their use to those who would appropriately benefit.
Tools to Help With SPECT Imaging of Cardiac Amyloidosis
There are several helpful resources available to help incorporate 99mTc-PYP imaging into clinical practice and nuclear laboratories. These efforts are led by the American Society of Nuclear Cardiology (ASNC), which offers several tools to educate physicians to better differentiate and diagnose ATTR cardiac amyloidosis. ASNC recently completed a three part webinar series on cardiac amyloidosis which is now archived on its website.
Additionally, ASNC has prepared and recently revised “Practice Points” on 99mTc-PYP Imaging for Transthyretin Cardiac Amyloidosis." This document provides an easily accessible fact sheet that summarizes important studies and clinical trials regarding ATTR CA and 99mTc-PYP imaging. Additionally, ASNC and several other medical societies are preparing an expert consensus document on multimodality imaging in cardiac amyloidosis. This highly anticipated document will be released later this year.
Noninvasive diagnosis of ATTR-CA represents an important new application of radionuclide imaging. Identification of its value in ATTR-CA diagnosis is timely given the emerging treatment options and recognition of the underdiagnosis of this morbid condition. Further research will refine use of these techniques and identify important new applications.
Editor’s note: Author Christopher A. Hanson, M.D., is a cardiology fellow specializing in advanced cardiac imaging at the University of Virginia. Author Jamieson M. Bourque M.D., MHS, is the medical director of nuclear cardiology and an associate professor of medicine and radiology at the University of Virginia.
References:
1. Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.

 January 05, 2023
January 05, 2023 

 imaging program. She spoke on this topic at the 2019 meeting of the American Society Nuclear Cardiology (ASNC) and led a tour with attendees of the PET-CT system at Rush, which was located close to the conference. #ASNC")