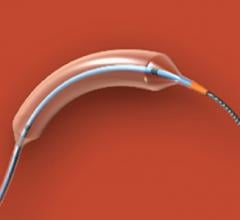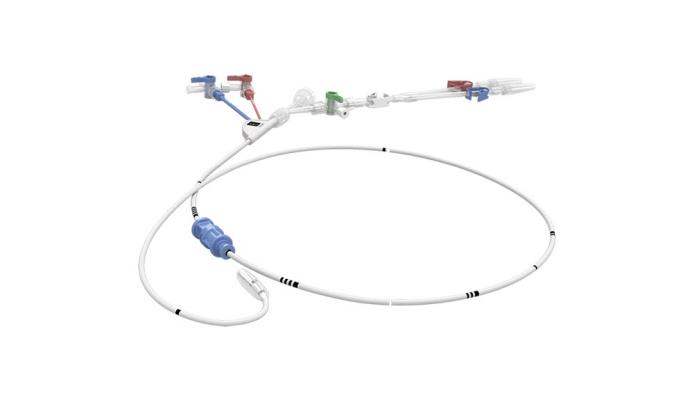
July 15, 2019 — Edwards LifeSciences is recalling the IntraClude Intra-Aortic Occlusion Device due to a risk of balloon rupture during use, which may add time to the procedure and compromises the safety of the patient.
The IntraClude Intra-Aortic Occlusion device is used in patients undergoing cardiopulmonary bypass, a technique in which a machine temporarily takes over the function of the heart and lungs during surgery. When the IntraClude balloon is inflated, the device blocks and vents (occludes) the aorta so that the heart is assessed without interference of other organs.
The IntraClude balloon bursting may cause serious adverse health consequences related to increased time the patient is on cardiopulmonary bypass, including neurological damage, embolism, stroke and death. Edwards has received 22 complaints related to balloon rupture or puncture. Three deaths have been reported.
On May 14, 2019, the company issued an Urgent Recall Notification sent to customers, listing the following actions:
-
Review the field safety notice to understand the potential hazard;
-
Complete and return the attached Acknowledgement Form within five business days of receiving this notice to Customer Service via fax at (800) 422-9329 or email to [email protected];
-
Complete and return the Product Reconciliation Form to Customer Service:
-
Record the quantity of any of the listed lot numbers in your possession
-
Contact Customer Service to arrange return and replacement of affected devices, and
-
Return affected devices to Edwards with the Return Goods Authorization (RGA) provided.
-
-
Distribute this notice within your organization or to any organization where the potentially affected devices have been transferred. If you have further distributed this product, notify your customers to the user level. Report any balloon failures to Edwards Lifesciences.
-
Your assistance is appreciated and necessary to ensure that this notice is reviewed, and that the response forms and affected devices are returned promptly.
Healthcare professionals and patients are encouraged to report adverse events or side effects related to the use of these products to the FDA's MedWatch Safety Information and Adverse Event Reporting Program:
-
Complete and submit the report online.
-
Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the form, or submit by fax to 1-800-FDA-0178.
The recall affects the IntraClude Intra-Aortic Occlusion Device model ICF100, UDI code (01) 00690103190007. All affected devices were distributed between May 1, 2017, and Feb. 19, 2019, encompassing a total of 757 devices in the U.S. Affected lot numbers include: 60972890, 61078031, 61097633, 61139239, 61259627, 61259628, 61713218, 61723505 and 61898939.
For more information: www.edwards.com
 June 13, 2024
June 13, 2024