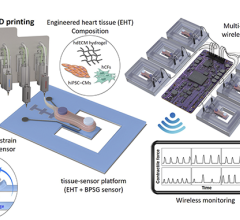
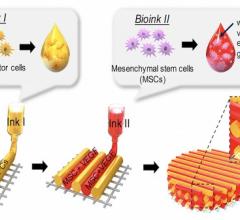
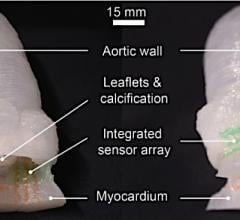
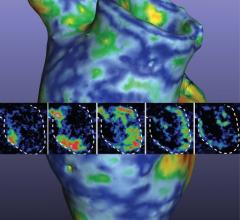
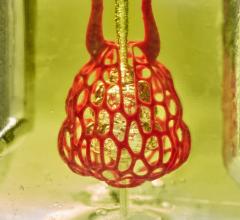
3-D printed vessel made from living cells implanted in an animal to test feasibility of using such structures clinically. Blood vessels are a stepping stone to the creation of more complex 3-D printed organs made from biomaterials. Image from the South Carolina Project for Organ Biofabrication.
The U.S. Food and Drug Administration (FDA) changed its rules concerning custom medical devices Oct. 12, adding new definitions to better outline what constitutes a device that does not require premarket or 510(k) marketed clearances, or requirements for clinical trials to measure its efficacy or safety. The FDA also released a guidance document for manufacturers and FDA staff regarding these amendments. The FDA made the changes as these types of devices are being eyed for guidance in the creation of custom, one-off, 3-D printed medical devices.
FDA said it has amended its regulations on the definition of a custom device so it includes new enumerated statutory requirements for custom devices under the Federal Food, Drug, and Cosmetic Act (FD&C) as amended by the Food and Drug Administration Safety and Innovation Act (FDASIA). This new provision amends the existing custom device exemption and introduces new concepts and procedures applicable to custom devices. The FDA said this action is being taken to align the regulations with the FD&C Act.
There is also growing interest in this exception to the FDA’s review of medical devices, as on-demand, custom 3-D printed medical devices are starting to be used more frequently. This rule has been cited by institutions that are already using 3-D printing (also called additive manufacturing) to create single, customized medical devices. In the case of 3-D printed devices made by a hospital to serve the needs of a specific patient, the hospital becomes the manufacturer that is governed by these rules.
Read the article “The Future of 3-D Printing in Medicine” to learn more about the current state and future applications of 3-D printing of medical devices.
Under the revised provision, as under the original custom device exemption, a device that meets the qualification of a custom device is exempt from 510(k) and premarket approval (PMA) submissions under sections 514 and 515 of the FD&C Act (21 U.S.C. 360d and 360e). The FDA said the current regulatory definition for a custom device was no longer consistent with the statute. This technical amendment will correct the regulations by revising the definition of a custom device to restate the statute.
The rule offers exemptions for manufacturers of custom devices that fit the needs of a particular patient. The reasoning is that it would be unreasonable to required manufacturers to file a premarket notification for each particular device.
The amendment to section 520(b) of the FD&C Act states that a device will qualify as a custom device by meeting new enumerated statutory requirements, including the following for each device:
1. It is created or modified in order to comply with the order of an individual physician or dentist.
2. It necessarily deviates from an otherwise applicable performance standard under section 514 or requirement under section 515 of the FD&C Act.
3. It is not generally available in the United States in finished form through labeling or advertising by the manufacturer, importer or distributor for commercial distribution.
4. It is designed to treat a unique pathology or physiological condition that no other device is domestically available to treat.
5. Either it is intended to meet the special needs of a physician or dentist in the course of the professional practice, or the device is intended for use by an individual patient named in the order by a physician or dentist.
6. The device is assembled from components or manufactured and finished on a case-by-case basis to accommodate the unique needs of and individual, physician or dentist.
7. It may have common, standardized design characteristics, chemical and material compositions, and manufacturing processes as commercially distributed devices.
The new provisions for the custom device exemption also include the following limitations:
1. The device is for the purpose of treating a sufficiently rare condition, such that conducting clinical investigations on such device would be impractical.
2. The production of the device must be limited to no more than five units per year of a particular device type.
3. A manufacturer is required to submit an annual report to FDA on the custom devices it supplied.
The FDA said this technical amendment to the regulations for the custom device exemption will ensure clarity and consistency with the requirements of the FD&C Act. Some manufacturers might be unaware that certain medical devices that they distribute as custom devices do not meet the statutory definition as currently described in the regulations and are subject to premarket review.
Also, FDA issued the final guidance entitled, “Custom Device Exemption Guidance for Industry and FDA Regulatory Staff,” explaining the new statutory provisions for custom devices. The guidance provides definitions of certain terms used in connection with the custom device exemption and explains how FDA interprets the devices that may qualify for the custom device exemption under section 520(b) of the FD&C Act. The guidance also describes in further detail what information should be submitted in an annual report, and provides recommendations on how to submit an annual report for custom devices distributed under the exemption. The guidance document can be found at www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddev-gen/documents/document/ucm415799.pdf.

 October 21, 2024
October 21, 2024