October 25, 2016 — St. Jude Medical is recalling 251,346 of its Fortify, Unify and Assura implantable cardioverter defibrillators (ICD) and cardiac resynchronization therapy defibrillators (CRT-D) because the batteries can deplete prematurely.
(Updated information on this recall -- In April 2017, the FDA issued a warning letter to Abbott / St. Jude Medical related to the company’s mismanagement of the recall, including sending recalled devices to hospitals that were implanted into patients. Read the article “FDA Harshly Criticizes Abbott, St. Jude For Failure to Address EP Device Safety.”)
St. Jude Medical is recalling certain models of the Fortify, Unify and Assura ICDs and CRT-Ds due to reports of rapid battery failure caused by deposits of lithium (known as lithium clusters) forming within the battery and causing a short circuit. If the battery unexpectedly runs out before the patient is aware of the rapid battery drain and able to have it replaced, the ICD or CRT-D will be unable to deliver life-saving pacing or shocks, which could lead to patient death.
The U.S. Food and Drug Administration (FDA) has identified this as a Class I recall, the most serious type of recall. The FDA said use of these devices may cause serious injuries or death.
St. Jude Medical reported that in some cases, full battery drainage can occur within a day to a few weeks after the patient receives an ERI alert. If the battery runs out, the ICD or CRT-D will be unable to deliver life-saving pacing or shocks, which could lead to patient death. The patients most at risk are those with a high likelihood of requiring life-saving shocks and those who are pacemaker dependent.
FDA and St. Jude Medical are alerting patients, patient-caregivers and physicians to respond immediately to elective replacement indicator (ERI) alerts. Due to problems with these batteries, patients do not have the normal three-month lead time for device replacement. Some batteries have run out within 24 hours of the patient receiving an ERI alert.
Scope of Battery Depletion Issue
To date, St. Jude said of the 398,740 affected devices sold worldwide, 841 were returned for analysis due to premature battery depletion caused by lithium clusters. Patient safety incidents related to this issue include:
• Two deaths (one in the U.S.) have been associated with devices that could not provide needed shock therapy due to premature battery depletion.
• Ten patients (nine in the U.S.) have reported fainting from devices that could not provide needed pacing therapy due to premature battery depletion.
• Thirty seven patients (30 in the U.S.) have reported dizziness from devices that could not provide needed pacing therapy due to premature battery depletion.
Battery depletion may not always be reported to the manufacturer, therefore the true number of devices with premature battery depletion due to lithium clusters is not known, St, Jude said. At this time, 349,852 affected devices remain actively implanted worldwide.
Affected ICD and CRT-D Devices
The recall affects the Fortify, Unify, and Assura ICD CRT-D devices manufactured between January 2010 to May 2015. These devices were distributed between February 2010 to October 2016. Devices recalled in the U.S. alone total 251,346 nationwide, the FDA said.
On Oct. 10, 2016, St. Jude Medical sent notification letters to customers and healthcare providers informing them of the possibility of premature battery depletion in affected ICD and CRT-D devices. Additionally, on Oct. 11, the FDA issued a safety communication regarding this recall, and provided recommendations for healthcare providers, patients and caregivers.
Full List of Affected Devices includes:
• Fortify VR: Model No(s). CD1231-40, CD1231-40Q
• Fortify ST VR: Model No(s). CD1241-40, CD1241-40Q
• Fortify Assura VR: Model No(s). CD1257-40, CD1257-40Q, CD1357-40C, CD1357-40Q
• Fortify Assura ST VR: Model No(s). CD1263-40, CD1263-40Q, CD1363-40C, CD1363-40Q
• Fortify DR: Model No(s). CD2231-40, CD2231-40Q.
• Fortify ST DR: Model No(s). CD2241-40, CD-2241-40Q, CD2263-40, CD2263-40Q
• Fortify Assura DR: Model No(s). CD2257-40, CD2257-40Q, CD2357-40C, CD2357-40Q
• Fortify Assura ST DR: Model No(s). CD2363-40C, CD2363-40Q
• Unify: Model No(s). CD3231-40, CD3231-40Q
• Unify Quadra: Model No(s). CD3249-40, CD3249-40Q
• Unify Assura: Model No(s). CD3257-40, CD3257-40Q, CD3357-40C, CD3357-40Q
• Quadra Assura: Model No(s). CD3265-40, CD3265-40Q, CD3365-40C, CD3365-40Q
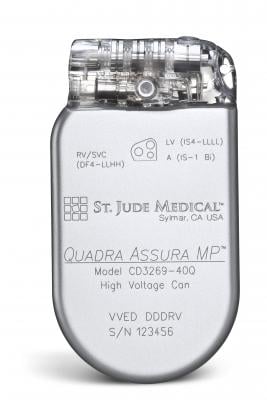
• Quadra Assura MP: Model No(s). CD3269-40, CD3269-40Q, CD3369-40C
Recommendations for Providers
• Do not implant unused affected devices. Premature battery depletion due to lithium clusters has only been observed in devices manufactured prior to May 2015. At this time, there is no information indicating that this issue affects devices manufactured after this date.
• Communicate with all patients who have an affected device that their device has a battery that may run out earlier than expected. Consider giving patients the Dear Patient letter provided by St. Jude Medical.
• Continue to conduct follow-up on patients with affected devices using in-office visits in addition to remote monitoring once they have been notified of the battery issue. Increased in-office surveillance is not necessary for patients who are also followed with remote monitoring.
• Immediately replace the device at the time of an ERI alert. Currently, there is not a factor, method or test to identify when devices with this form of premature battery depletion are approaching ERI, or to accurately predict remaining battery life once ERI appears.
• Pacemaker-dependent patients with a device that has reached ERI should be treated as a medical emergency.
• Healthcare providers should consider whether elective device replacement is warranted for their pacemaker dependent patients. Ultimately, health care providers should individualize the care of their patients based on the patients’ medical history comorbidities and condition.
• Most patients will not require prophylactic device replacement prior to ERI, as the rate of complications following replacement surgery are higher than those associated with premature battery depletion. However, the FDA and St. Jude Medical recognize the need to weigh individual clinical considerations. If the decision is made to replace an affected device based on individual patient circumstances, St. Jude Medical has announced they will provide a replacement device at no cost.
• Enroll patients in Merlin@Home, St. Jude Medical’s home monitoring system for these devices, especially those who have difficulty recognizing their device’s ERI alerts. For patients already enrolled in Merlin@Home, explain the importance of ongoing home monitoring. Utilize the “Direct Alerts” feature to provide you with an alert notification when a patient’s device has reached ERI. Please see additional information about the Merlin@Home Monitoring System below. If a home monitor is ordered for a patient with an affected device, St. Jude Medical will cover the cost of the home monitor.
• Ensure that the ERI battery alert is ON for all patients. Review the most recent “Programmed Parameters” printout.
• Review the “Trigger Alerts When” section, and ensure that the “Device at ERI” parameter is “on” for both the “Show on FastPath” and “Notify Patient” selections.
• If the “Device at ERI” alert is “off”, the patient should be seen promptly to program this parameter to “on”.
• Advise affected patients that an ERI alert triggers a vibratory notification and perform the following procedures at each scheduled office visit:
1. Interrogate the patient’s device to determine if an ERI alert has been triggered. Premature battery depletion can be identified by health care providers through home monitoring showing ERI or more advanced battery depletion.
2. Perform a patient notifier test to confirm that the patient feels and recognizes the vibratory alert.
3. Patients who cannot feel the vibratory alert may experience loss of battery and/or loss of device function without their awareness. Advise the patient to contact your office promptly should they feel a vibratory alert. An in-office evaluation should be performed to determine the reason for the alert as other non-critical events can also trigger a vibratory alert.
St. Jude Contact and Additional Information
Customers with questions may contact St. Jude Medical Customer Service at (866) 915-5065.
Read more information from St. Jude Medical on the premature battery depletion issue
Read the Global Medical Advisory Concerning Subset of ICD and CRT-D Devices
Read the FDA’s safety communication

 July 21, 2025
July 21, 2025