May 8, 2024 — The US Food and Drug Administration (FDA) is alerting health care providers and facilities about our continued safety and quality concerns with the following Getinge/Maquet cardiovascular medical devices:
- Getinge/Maquet/Datascope Cardiosave Hybrid and Rescue Intra-Aortic Balloon Pump (IABP) devices and
- Getinge/Maquet Cardiohelp system and HLS Sets.
The FDA recommends that health care providers and facilities transition away from these devices and seek alternatives, if possible. These recommendations are based on our continued concerns that Getinge/Maquet has not sufficiently addressed the problems and risks with these recalled devices.
The FDA continues to receive medical device reports (MDR) related to the problems even though Getinge/Maquet took corrective steps, including reinforcing information in the Instructions for Use as well as providing users with new actions they should take when using the IABP.
The FDA recognizes that alternative treatment options are limited and has provided the recommendations below for health care providers when these Getinge cardiovascular devices are used.
Recommendations
- Plan for alternative capital equipment to transition away from these Getinge cardiovascular devices:
- Getinge/Maquet/Datascope Cardiosave Hybrid and Rescue Intra-Aortic Balloon Pump (IABP) devices.
- Getinge Cardiopulmonary bypass (CPB) devices including the Getinge/Maquet Cardiohelp system and HLS Sets.
- Use alternative devices, if possible. If you don’t have alternatives and continue to use these devices:
- Review the FDA’s previous recommendations. Read any Urgent Medical Device Correction notices from Getinge and follow the recommendations.
- Be aware of the recalls related to these devices.
- Report any issues or adverse events with Getinge devices to the FDA. For details on reporting, see Reporting Problems to the FDA.
- Report any supply chain issues to the [email protected] mailbox.
Background
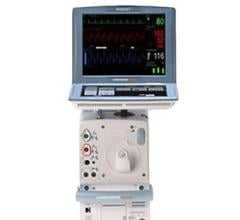
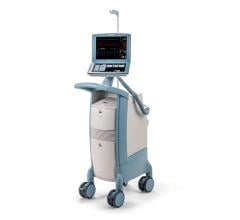
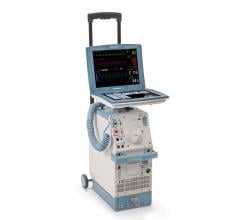
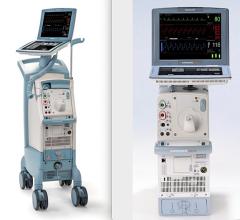
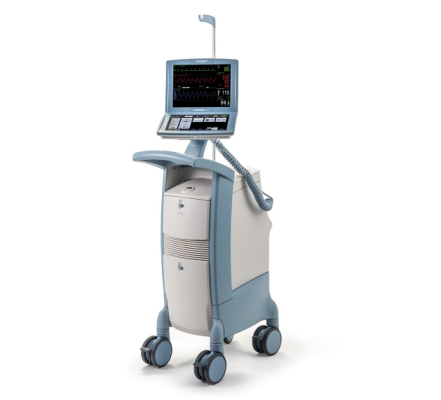
Cardiosave Hybrid and Rescue IABP
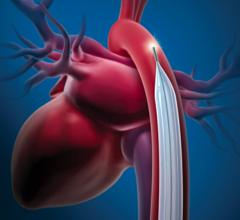
The Getinge/Maquet/Datascope Cardiosave Hybrid and Rescue IABP is a cardiac assist device placed in the artery that is in the chest and abdomen (descending aorta). The device is an electromechanical system used to inflate and deflate intra-aortic balloons. These systems provide temporary support to the heart's left ventricle through counter pulsation.
-
Cardiosave Recalls and MDRs
- From January 1, 2023 through April 11, 2024, Getinge initiated 12 voluntary recalls in the U.S. for the Cardiosave IABP. Of these, the FDA classified eight as a Class I recall, the most serious type of recall. In the last 12 months, the FDA has received 2,964 medical device reports (MDR) related to Cardiosave Intra-Aortic Balloon Pumps. Of those, 15 were reported as resulting in patient serious injury or death. As a general matter, MDRs are one of the FDA’s several postmarket surveillance tools used to monitor device performance, detect potential device-related safety issues, and contribute to benefit-risk assessments during real world use. It may be difficult to confirm a direct cause and effect between an adverse event report and a specific medical device based on the limited information provided in the reports.
- The FDA continues to receive adverse event reports related to the failures associated with Cardiosave recalls. Since 2017, the FDA has been evaluating and monitoring MDRs that describe Cardiosave IABP devices shutting down. The FDA has also been evaluating other concerns with the Cardiosave IABP, including blood entering the device, which can cause the IABP to rupture (helium emboli) or the patient or health care provider being exposed to patient blood.
- In addition, on March 1, 2024External Link Disclaimer, TÜV SÜD, the European Union notified body for certification services, temporarily suspended the CE certificate (CE marking means that products have Conformité Européenne or European conformity certification) for Getinge’s Cardiosave Intra-Aortic Balloon Pump with immediate effect. Getinge is given six months to adhere to the required corrective actions, and during this time, the Intra-Aortic Balloon Pumps cannot be sold in countries that require CE certification. This only impacts the Cardiosave devices.
Cardiohelp system and HLS Sets
The Getinge/Maquet Cardiohelp system is a cardiopulmonary bypass device that pumps blood out of the patient to oxygenate the blood during cardiopulmonary bypass surgeries. It can be used for up to six hours. The HLS Set is an oxygenator and blood pump, which is a disposable component of the Cardiohelp system.
-
Cardiohelp and HLS Set Recalls and MDRs
- From January 1, 2023, through April 11, 2024, Getinge initiated eight voluntary recalls for the Cardiohelp system, including the HLS Set. Of these, the FDA classified one as a Class I recall, the most serious type of recall. In the last 12 months, the FDA received 246 MDRs related to the Getinge Cardiohelp System, including the HLS Set. Of those, 33 were reported as resulting in patient serious injury or death. However, as stated above it may be difficult to confirm a direct cause and effect between an adverse event report and a specific medical device based on the limited information provided in the reports. The FDA continues to have concerns regarding the sterility of the HLS Set.
- Sterility was also a concern in May 2023, when Getinge notified U.S. customers that it removed another of its cardiopulmonary bypass devices, the Quadrox oxygenators, from the U.S. market. The Quadrox oxygenators continue to be unavailable because of sterility issues.
FDA Actions
The FDA continues to work with the Getinge to understand factors contributing to the device failures, as well as possible mitigation strategies.
The FDA is working with other manufacturers to assess their interest and ability to manufacture and distribute alternative devices in the U.S.
The FDA worked with the U.S. Department of Justice to place Getinge manufacturing sites under consent decree in 2015 and added the IABP manufacturing site in 2022. This action allows additional FDA oversight, an independent auditor, inspections, and updates on progress made toward addressing quality and safety concerns. At this time, the cardiopulmonary bypass and IABP facilities have not met the requirements to have the consent decree lifted.
The FDA will continue to keep health care providers and the public informed if new or additional information becomes available.
Previous FDA Communications
- UPDATE: Risk of Device Failures for Getinge’s Maquet/Datascope Cardiosave Intra-Aortic Balloon Pump (IABP) – Letter to Health Care Providers
- Getinge/Maquet Cardiohelp System: Potential Insufficient Packaging Sterility with HLS Set Advanced - Letter to Health Care Providers
- Oxygenator Devices Used for Extracorporeal Circulation - Letter to Health Care Providers
Unique Device Identifier (UDI)
The FDA established the unique device identification system to adequately identify medical devices sold in the United States from manufacturing through distribution to patient use. For more information on UDI, please visit Unique Device Identification System (UDI Systems).
Reporting Problems to the FDA
If you are experiencing supply issues, contact the FDA about a medical device supply chain issue. Reporting supply issues informs the FDA of how it may be able to help address device supply availability.
The FDA encourages health care providers to report any adverse events or suspected adverse events experienced with the Getinge devices.
- You can submit voluntary reports through MedWatch, the FDA Safety Information and Adverse Event Reporting program.
- Device manufacturers and user facilities must comply with the applicable Medical Device Reporting (MDR) regulations.
- Health care personnel employed by facilities that are subject to the FDA's user facility reporting requirements should follow the reporting procedures established by their facilities.
By promptly reporting adverse events, you can help the FDA identify and better understand the risks associated with medical devices.
Contact Information
If you have questions about this letter, contact the Division of Industry and Consumer Education (DICE).
 August 14, 2023
August 14, 2023