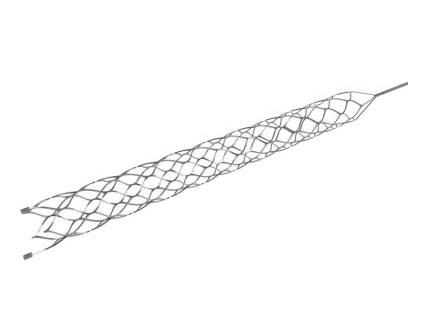
May 21, 2018 — Medtronic and the U.S. Food and Drug Administration (FDA) announced a Class I Urgent Medical Device Recall of the MindFrame Capture LP revascularization device on account of a risk of the delivery wire breaking or separating during use. The FDA said the clot retriever could be left inside the patient’s bloodstream, and this or the attempts made to retrieve the device, can lead to further complications; these complications include bleeding, additional blockage of blood vessels, more severe stroke symptoms or death.
On April 4, 2018, Medtronic followed up with their customers with another Urgent Medical Device Recall notice regarding patient management. The notice recommended healthcare providers to:
- Review the notification and distribute the information to all appropriate personnel;
- Follow up with the patient closely;
- Consider antiplatelet therapy;
- Consider repeating imaging on the patient; and
- Complete and return the acknowledgement and receipt form to Medtronic.
The MindFrame Capture LP revascularization device is intended to restore blood flow or remove blood clots within a blood vessel in the brain during an acute ischemic stroke in patients who are ineligible for or fail intravenous tissue plasminogen activator (IV t-PA) therapy.
The total number of devices recalled in the U.S. has reached 529, according to the FDA. Product lot numbers affected by the recall are as follows: 300010, 300011, 300012, 300013, 300014, 300015, 300016, 300017 and 300018. Affected devices were manufactured between Feb. 3, 2016, and Jan. 14, 2018; they were distributed between March 18, 2016 and Jan. 17, 2018.
Customers with questions may contact Medtronic Quality Assurance by email at [email protected] or by phone at 1(800) 633-8766.
Healthcare professionals and patients are encouraged to report adverse events or side effects related to the use of these products to the FDA's MedWatch Safety Information and Adverse Event Reporting Program:
- Complete and submit the report online: www.fda.gov/MedWatch/report
- Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
For more information: www.medtronic.com, www.fda.gov

 May 29, 2026
May 29, 2026 







