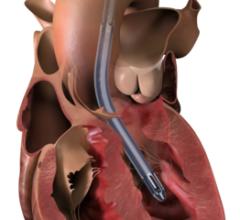
April 11, 2016 — Abiomed Inc. announced that it has received U.S. Food and Drug Administration (FDA) pre-market approval (PMA) for its Impella heart pumps to provide treatment of ongoing cardiogenic shock. In this setting, the pumps — which include the Impella 2.5, Impella CP, Impella 5.0 and Impella LD — stabilize the patient's hemodynamics, unload the left ventricle, perfuse the end organs and allow for recovery of the native heart.
This latest approval adds to the prior FDA indication of Impella 2.5 for high risk percutaneous coronary intervention (PCI), or Protected PCI, received in March 2015.
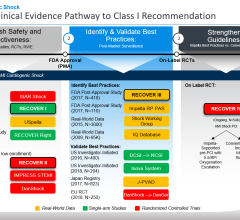
With this approval, these are the first and only percutaneous temporary ventricular support devices that are FDA-approved as safe and effective for the cardiogenic shock indication, as stated below:
The Impella 2.5, Impella CP, Impella 5.0 and Impella LD catheters, in conjunction with the Automated Impella Controller console, are intended for short-term use (<4 days for the Impella 2.5 and Impella CP and <6 days for the Impella 5.0 and Impella LD) and indicated for the treatment of ongoing cardiogenic shock that occurs immediately (<48 hours) following acute myocardial infarction (AMI) or open heart surgery as a result of isolated left ventricular failure that is not responsive to optimal medical management and conventional treatment measures with or without an intra-aortic balloon pump. The intent of the Impella system therapy is to reduce ventricular work and to provide the circulatory support necessary to allow heart recovery and early assessment of residual myocardial function.
The product labeling also allows for the clinical decision to leave Impella 2.5, Impella CP, Impella 5.0 and Impella LD in place beyond the intended duration of four to six days due to unforeseen circumstances.
The Impella products offer the ability to both stabilize the patient's hemodynamics before or during a PCI procedure and unload the heart, which allows the muscle to rest and potentially recover its native function. Heart recovery is the ideal option for a patient's quality of life and, as documented in several clinical papers, has the ability to save costs for the healthcare system.
Cardiogenic shock is a life-threatening condition in which the heart is suddenly unable to pump enough blood and oxygen to support the body's vital organs. For this approval, it typically occurs during or after a heart attack or AMI or cardiopulmonary bypass surgery as a result of a weakened or damaged heart muscle. Despite advancements in medical technology, critical care guidelines and interventional techniques, AMI cardiogenic shock and post-cardiotomy cardiogenic shock (PCCS) carry a high mortality risk and has shown an incremental but consistent increase in occurrence in recent years in the United States.
"This approval sets a new standard for the entire cardiovascular community as clinicians continue to seek education and new approaches to effectively treat severely ill cardiac patients with limited options and high mortality risk," said William O'Neill, M.D., medical director of the Center for Structural Heart Disease at Henry Ford Hospital. "The Impella heart pumps offer the ability to provide percutaneous hemodynamic stability to high-risk patients in need of rapid and effective treatment by unloading the heart, perfusing the end organs and ultimately, allowing for the opportunity to recover native heart function."
The data submitted to the FDA in support of the PMA included an analysis of 415 patients from the RECOVER 1 study and the U.S. Impella registry (cVAD Registry), as well as an Impella literature review including 692 patients treated with Impella from 17 clinical studies. A safety analysis reviewed over 24,000 Impella treated patients using the FDA medical device reporting ("MDR") database, which draws from seven years of U.S. experience with Impella.
In addition, the company also provided a benchmark analysis of Impella patients in the real-world Impella cVAD registry versus these same patient groups in the Abiomed AB5000/BVS 5000 Registry. The Abiomed BVS 5000 product was the first ventricular assist device (VAD) ever approved by the FDA in 1991 based on 83 patient PMA study. In 2003, the AB5000 Ventricle received FDA approval and this also included a PMA study with 60 patients.
For this approval, the data source for this benchmark analysis was a registry ("AB/BVS Registry") that contained 2,152 patients that received the AB5000 and BVS 5000 devices, which were originally approved for heart recovery. The analysis examined by the FDA used 204 patients that received the AB5000 device for the same indications. This analysis demonstrated significantly better outcomes with Impella in these patients.
Watch a VIDEO demonstration of how the Impella works.
For more information: www.abiomed.com
 April 28, 2023
April 28, 2023