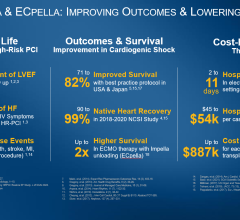
The Abiomed Impella RP had higher than expected mortality in its post-approval study, much higher than in its pre-market approval study. The vendor and the FDA believe this might be due to poor patient selection and implanting the device too late to aid the patient.
The U.S. Food and Drug Administration (FDA) sent a letter to cardiologists this week to explain its evaluation of high mortality rates reported in the Abiomed Impella RP heart pump post-approval study (PAS). The FDA believes the mortality rate might be higher than the pre-market study because of inappropriately selected patients receiving the device.
"Although the FDA is concerned about the high mortality rate from the interim PAS results, we believe that when the device is used for the currently approved indication in appropriately selected patients, the benefits of the Impella RP system continue to outweigh the risks," said William Maisel, M.D., MPH, chief medical officer for the FDA Center for Devices and Radiological Health, in the letter to providers. "We do not know the root cause for the high mortality rate, and the results (of the PAS) are not adjusted for potential confounders."
In the premarket clinical studies, where strict inclusion and exclusion criteria were followed, a total of 44 out of 60 patients (73.3 percent) survived to 30 days. This included post-device explant, hospital discharge (whichever was longer), or to the start of longer-term therapy such as heart transplant or implantation of a surgical right ventricular assist device (RVAD).
The FDA mandated manufacturer of the device, Abiomed, to conduct a PAS as a condition of approval for the Impella RP System. The study follows 60 newly treated patients for one year through the firm’s cVAD registry. The primary endpoint is survival to 30 days post device explant or hospital discharge (whichever is longer), or to the start of next longer-term therapy.
Interim results from the most recent PAS report, which reflect device use in a broader patient population, indicate that only four out of the 23 enrolled PAS patients (17.4 percent) met the primary survival endpoint.
The FDA required additional analyses from Abiomed, and data submitted by the company in January 2019 suggests that the high mortality rate observed might be related to differences in pre-implant characteristics of the PAS patients compared to the patients in the premarket clinical studies. Sixteen of the 23 patients enrolled in the PAS would not have met the enrollment criteria for the premarket clinical studies.
Specifically, before getting the Impella RP system implanted, patients in the PAS were more likely than the premarket clinical study patients to have been in cardiogenic shock for longer than 48 hours, experienced an in-hospital cardiac arrest, been treated with an intra-aortic balloon (IABP) pump, or suffered a pre-implant hypoxic or ischemic neurologic event.
The temporary, percutaneous right ventricular heart pump is designed to provide right side hemodynamic support. It is intended to help patients maintain stable heart function without open chest surgery. The FDA approved the Impella RP System in September 2017. The device is implanted centrally via peripheral access to help patients who require temporary emergency support of right ventricular function. Use of the device, which may be up to 14 days, requires patients to stay in the hospital.
Abiomed Issues Safety Bulletin on Impella RP
Abiomed issued its own safety bulletin to users in January and said the issue in the PAS data appears to be due to patient selection. The company said the use on some patients did not follow the proper inclusion/exclusion criteria set up by the FDA and the company. Implant timing also appeared to be an issue, Abiomed stated, with the device not being used early enough to aid the patient.
The company said the FDA studies demonstrated that the proper use and timely implantation of the Impella RP yields higher survival with the potential for recovery of the right ventricle. These protocols are captured in multiple physician and hospital publications, and identified as best practices.
The company tracks commercial use of all its percutaneous pumps in its Impella Quality Database (IQ). The vendor is also conducting the cVAD Study at approximately 30 hospitals utilizing IRB and prospective data collection with FDA definitions from prior studies.
Looking at the most recent PAS data, Abiomed said of the 23 patients in the PAS cohort, only 7, or 30 percent, would have met the criteria from the FDA study. Additionally 70 percent qualify as a salvage attempt or last resort implantation. The table below shows the statistical differences in the patient characteristics or timing of treatment:
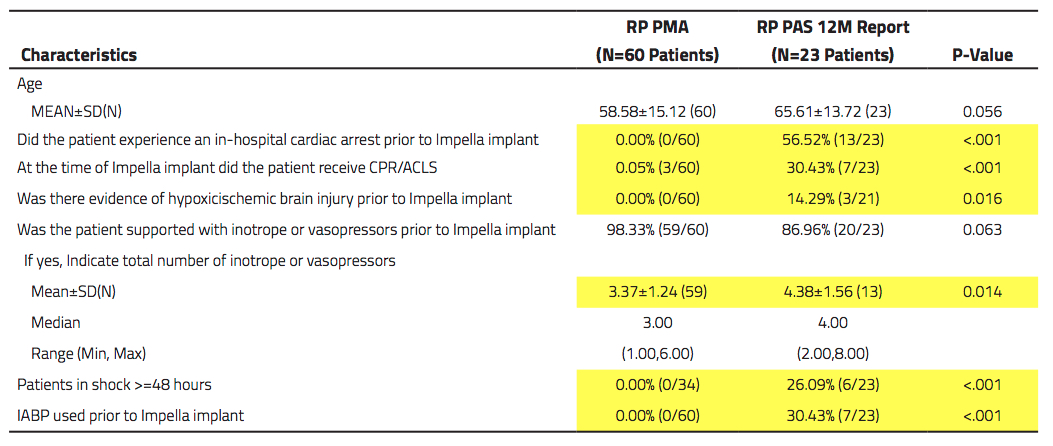
The dominant change that was observed in the PAS was prolonged time in shock (more than 24 hours) before placement of the Impella RP device in these patients, Abiomed stated. This review of patients in cVAD detailed frequent use of the device after periods of RV failure of more than 48 hours, up to several weeks post-LVAD implant, post-open heart surgery, or post-myocardial infarction with cardiogenic shock. Abiomed strongly recommends adherence and compliance to the inclusion/exclusion criteria used in the PMA cohort to ensure optimal outcomes.
Abiomed said conversations about the risks should take place with patients and their families, but also should be extended prior to the use of alternative devices, including ECMO, TandemLife or Centrimag. The company noted these devices were not subject to post-approval studies or FDA reporting.
Abiomed developed a patient selection checklist for the Impella RP, which is accessible at www.protectedpci.com/wp-content/uploads/2019/02/IMP-574_Impella_RP-Patient-Selection_checklist.pdf.
FDA Recommendations for Use of the Impella RP
The FDA has the following recommendations for healthcare providers:
• Be aware that FDA approval of the Impella RP System was based on the results of premarket clinical studies that included patients who had been in cardiogenic shock for less than 48 hours prior to device implant. Additionally, none of the patients in the premarket clinical studies experienced an in-hospital cardiac arrest, or were treated with an IABP, or suffered a hypoxic or ischemic neurologic event, prior to Impella RP being implanted. Although these clinical events may not preclude a clinical decision to use the device, physicians should be aware that the occurrence of one or more of these events prior to Impella RP implantation may decrease expected survival rate.
• Carefully consider these interim survival results from the ongoing PAS when making treatment decisions and discuss the risks and benefits of the Impella RP System with patients and their caregivers. Additionally, be aware that there are currently no other device interventions that have been approved by the FDA under the premarket application (PMA) process for the patient population demonstrating a higher mortality rate in the PAS and as such, other interventions pose risks, as well, that should be considered and discussed with patients and their caregivers.
• Report any adverse events or suspected adverse events experienced with the Impella RP. Voluntary reports can be submitted through MedWatch, the FDA safety information and adverse event reporting program.
• Device manufacturers and user facilities must comply with the applicable medical device reporting (MDR) regulations. Prompt reporting of adverse events can help the FDA identify and better understand the risks associated with medical devices.
FDA Safety Review of Impella is Ongoing
The FDA said it will continue to review data from the ongoing PAS and other available data sources as they become available. The FDA will work with Abiomed to ensure the product labeling addresses the PAS interim results.
Read the October 2019 update article "FDA Post Clearance Study Shows Timely Diagnosis of Right Heart Failure and Early Impella RP Use Leads to Better Survival."
Read the March 2019 update article "Right Heart Hemodynamic Support With Impella RP, Getting it Right."
Related Impella RP Content:
Abiomed Receives FDA PMA Approval for Impella RP for Right Heart Failure
Impella RP Clinical Trial Results Released
FDA Grants HDE for Abiomed’s Impella RP for Right Ventricular Support
ECRI Institute Review Supports Impella RP Device
UH Case Medical Center First in Ohio to Implant Impella RP Ventricular Assist Device
First Patient Supported with Transcatheter Right Heart VAD
Advances in Percutaneous Ventricular Assist Devices
Beaumont Hospital Offers New Device for Right-Side Heart Failure Patients
Abiomed Surpasses 15,000 Impella Patients in the United States
Abiomed Showcases Expanded FDA Indications, Next-Generation Heart Recovery Products at TCT 2018
Trends and Advances in Hemodynamic Support
 April 28, 2023
April 28, 2023